Simulierter Verschleiß
Computersimulationen zeigen Verschleiß und Reibung realer Werkstoffe auf atomarer Ebene.
Verschleiß und Reibung sind ganz entscheidende Themen für viele Industriebereiche: Was passiert, wenn eine Oberfläche über eine andere gleitet? Mit welchen Materialveränderungen muss man dabei rechnen? Was bedeutet das für die Haltbarkeit und Sicherheit von Maschinen? Was dabei auf atomarer Ebene passiert, lässt sich nicht direkt beobachten. Nun steht dafür allerdings ein neues zusätzliches wissenschaftliches Werkzeug zur Verfügung: Aufwändige Computersimulationen werden nun erstmals so leistungsfähig, dass man Verschleiß und Reibung realer Werkstoffe auf atomarer Skala simulieren kann.
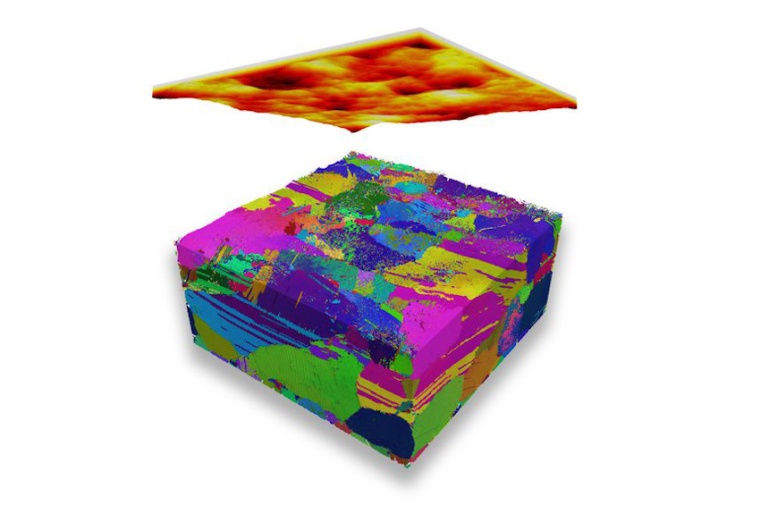
Dass diese neue Forschungsrichtung nun verlässliche Ergebnisse liefert, beweist das Tribologie-Team an der TU Wien, geleitet von Carsten Gachot. Das Verhalten von Oberflächen aus Kupfer und Nickel konnte mit großen Hochleistungsrechnern simuliert werden. Die Ergebnisse stimmen verblüffend genau mit Bildern aus dem Elektronenmikroskop überein – liefern aber noch wertvolle Zusatzinformation. Mit bloßem Auge sieht es nicht besonders spektakulär aus, wenn zwei Oberflächen aneinander gleiten. Doch auf mikroskopischer Ebene laufen dabei hochkomplizierte Vorgänge ab: „Metalle, wie man sie in der Technik verwendet, haben eine spezielle Mikrostruktur“, erklärt Stefan Eder. „Sie bestehen aus kleinen Körnchen, mit einem Durchmesser in der Größenordnung von Mikrometern oder noch kleiner.“
Wenn nun ein Metall unter großer Scherbelastung über das andere gleitet, dann geraten die Körnchen der beiden Materialien aneinander: Die Körnchen können dabei gedreht, verformt oder verschoben werden, sie können in kleinere Körnchen zerteilt werden oder durch erhöhte Temperatur oder mechanische Einwirkung wachsen. All diese Prozesse, die auf mikroskopischer Skala ablaufen, bestimmen letztlich das Verhalten des Materials auf großer Skala – und damit entscheiden sie auch über die Lebensdauer einer Maschine, wie viel Energie in einem Motor durch Reibung verlorengeht oder wie gut eine Bremse funktioniert, in der eine möglichst hohe Reibkraft erwünscht ist.
„Das Ergebnis dieser mikroskopischen Prozesse kann man danach unter dem Elektronenmikroskop untersuchen“, sagt Eder. „Man erkennt, wie sich die Kornstruktur der Oberfläche verändert hat. Allerdings war es bisher nicht möglich, den Ablauf dieser Prozesse zu studieren und genau zu erklären, wodurch welche Effekte zu welchem Zeitpunkt verursacht werden.“ Diese Lücke schließen nun große Molekulardynamik-Simulationen, die das Tribologie-Team in Zusammenarbeit mit dem Exzellenzzentrum für Tribologie (AC²T) in Wiener Neustadt und dem Imperial College in London& entwickelt: Atom für Atom werden die Oberflächen am Computer simuliert. Je größer das simulierte Materialstück und je länger der simulierte Zeitabschnitt, umso mehr Computerleistung wird benötigt. „Wir simulieren Abschnitte mit einer Seitenlänge von bis zu 85 Nanometern, über einige Nanosekunden hinweg“, sagt Eder.
Das Team untersuchte den Verschleiß einer Legierung aus Kupfer und Nickel bei unterschiedlichen Mischungsverhältnissen der beiden Metalle und unterschiedlichen mechanischen Belastungen. „Unsere Computersimulationen ergaben genau die Vielfalt an Prozessen, an Kornveränderungen und Verschleiß-Effekten, wie man sie aus Experimenten grundsätzlich bereits kennt“, sagt Eder. „Wir können damit Bilder produzieren, die genau den Aufnahmen aus dem Elektronenmikroskop entsprechen. Allerdings hat unsere Methode einen entscheidenden Vorteil: Wir können den Prozess danach am Computer im Detail analysieren. Wir wissen, welches Atom zu welchem Zeitpunkt seinen Platz gewechselt hat, und was mit welchem Körnchen in welcher Phase des Prozesses genau passiert ist.“
In der Industrie stoßen die neuen Methoden bereits auf großes Interesse. „Schon seit Jahren wird darüber diskutiert, dass die Tribologie von verlässlichen Computersimulationen profitieren könnte. Nun haben wir ein Stadium erreicht, in dem die Qualität der Simulationen und die verfügbare Rechenleistung so groß sind, dass wir dadurch spannende Fragen beantworten könnten, die auf andere Weise gar nicht zugänglich wären“, sagt Carsten Gachot. So möchte man in Zukunft auch industrielle Prozesse auf atomarer Ebene analysieren, verstehen und verbessern.
TU Wien / JOL
Weitere Infos
- Originalveröffentlichung
S. J. Eder et al.: Unraveling and Mapping the Mechanisms for Near-Surface Microstructure Evolution in CuNi Alloys under Sliding, ACS Appl. Mater. Interfaces, online 16. Juni 2020; DOI: 10.1021/acsami.0c09302 - Tribologie, Institut für Konstruktionswissenschaften und Produktentwicklung, Technische Universität Wien








