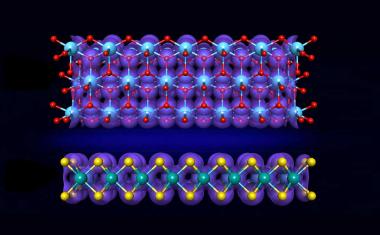
Eigenschaften von Kristall-Oberflächen automatisch vorhersagen
Neue Methode kann die Suche nach Materialien für Technologien wie Photovoltaik, Batterien und Datenübertragung beschleunigen.
Bei der Suche nach neuen Materialien für wichtige Technologien wie etwa Photovoltaik, Batterien oder Datenübertragung werden computergestützte Methoden immer wichtiger. Jetzt haben Caterina Cocchi und Holger-Dietrich Saßnick von der Uni Oldenburg ein Verfahren entwickelt, mit dem sich die physikalischen Eigenschaften komplexer Kristalloberflächen automatisiert und allein anhand grundlegender physikalischer Gesetzmäßigkeiten berechnen lassen. Das ermögliche es, schneller passende Materialien für Anwendungen, etwa aus dem Energiebereich, zu finden, so die Forscher. In Zukunft möchten sie ihr Verfahren mit künstlicher Intelligenz und den Möglichkeiten des maschinellen Lernens kombinieren, um den Prozess noch weiter zu beschleunigen.
Ähnliche Verfahren haben sich bislang auf massive Festkörper konzentriert und nicht auf Oberflächen. „Alle Prozesse, die wichtig sind, um Energie umzuwandeln, zu produzieren oder zu speichern, spielen sich aber auf Oberflächen ab“, so Cocchi. Es sei allerdings wesentlich schwieriger, Materialeigenschaften von Oberflächen zu berechnen als von vollständigen Kristallen. Die Grenzflächen sind meist komplex aufgebaut, Ursache dafür können beispielsweise Defekte in der Kristallstruktur oder ein ungleichmäßiges Wachstum eines Kristalls sein.
Diese Komplexität stellt Materialwissenschaftler vor Probleme: „Häufig lassen sich die Eigenschaften von Proben experimentell nicht eindeutig ermitteln“, sagt Cocchi. Das habe Saßnick und sie motiviert, ein automatisiertes Verfahren zu entwickeln, um die Charakteristika neuer Verbindungen mit hoher Qualität zu errechnen.
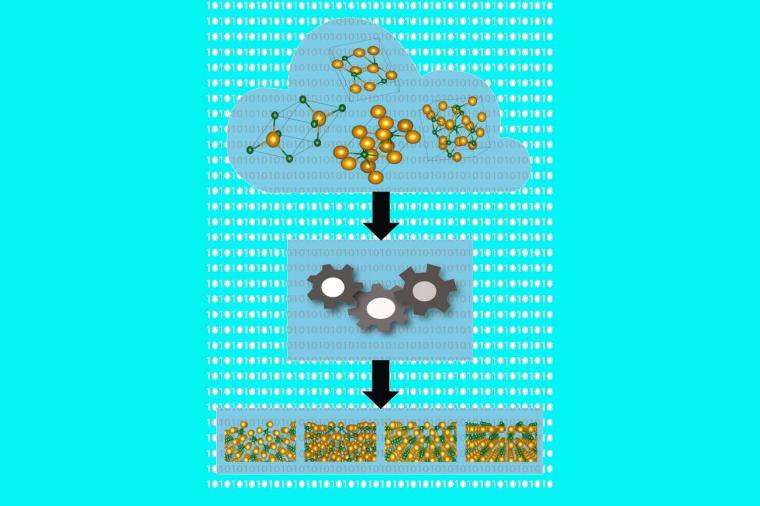
Das Ergebnis ihrer Arbeit ist Bestandteil eines Computerprogramms mit dem Namen „aim2dat“, das als Eingabe lediglich die chemische Zusammensetzung einer Verbindung benötigt. Die Kristallstruktur wird aus existierenden Datenbanken entnommen. Anschließend errechnet die Software zunächst, unter welchen Bedingungen die Oberfläche des Materials chemisch stabil ist. In einem zweiten Schritt ermittelt das Programm wichtige Eigenschaften, insbesondere, welche Energie nötig ist, um Elektronen in Leitungszustände anzuregen oder gar von der Oberfläche zu lösen.
Diese Größe spielt etwa in Materialien, die Sonnenenergie in elektrischen Strom umwandeln sollen, eine wichtige Rolle. „In unsere Berechnungen fließen keine Vorannahmen ein, sondern wir nutzen allein die fundamentalen Gleichungen der Quantenmechanik, weshalb unsere Ergebnisse sehr zuverlässig sind“, erläutert Cocchi.
Die Anwendbarkeit des Verfahrens demonstrierten die beiden Forscher am Beispiel des Halbleiters Cäsiumtellurid. Die Kristalle dieses Materials, das in Teilchenbeschleunigern als Elektronenquelle verwendet wird, können in vier unterschiedlichen Formen auftreten. „Die Zusammensetzung und Qualität von Proben des Materials sind in Experimenten nur schwer zu kontrollieren“, berichtet Saßnick. In ihren Berechnungen konnten die Forscher jedoch wichtige physikalische Eigenschaften detailliert für die verschiedenen Konfigurationen der Cäsiumtellurid-Kristalle ermitteln.
Cocchi und Saßnick haben die Software in eine öffentlich verfügbare Programmbibliothek eingebettet, damit auch andere Forscher die Möglichkeit haben, das Verfahren zu nutzen und zu verbessern. „Unsere Methode hat großes Potenzial, um neue Materialien für verschiedenste Anwendungen im Energiebereich zu entdecken – insbesondere physikalisch und strukturell komplex aufgebaute Festkörper“, sagt Cocchi.
U. Oldenburg / RK