Wenn Moleküle einen Reifenabdruck hinterlassen
Eine Kombination aus statistischer Physik und Simulationen mit rastertunnelmikroskopischen Aufnahmen soll die molekulare Selbstorganisation optimieren.
Wenn Moleküle einen Reifenabdruck hinterlassen
Eine Kombination aus statistischer Physik und Simulationen mit rastertunnelmikroskopischen Aufnahmen soll die molekulare Selbstorganisation optimieren.
Manche Moleküle können sich auf Oberflächen in spezifischen Mustern selbst anordnen. Diese Selbstorganisation spielt bei technischen Anwendungen eine Rolle, die auf funktionellen Oberflächenstrukturen beruhen. Bislang konnten diese Prozesse aber kaum gesteuert oder vorhergesagt werden. Einem Forscherteam vom "Center for NanoScience" (CeNS) der LMU München gelang hier nun ein Durchbruch: Eine Kombination aus statistischer Physik und detaillierten Simulationen mit rastertunnelmikroskopischen Aufnahmen lieferte ein vereinfachtes Modell zur Vorhersage der Muster. "Wir können nun in der Theorie eine Vielzahl von Mustern reproduzieren, die überraschend gut mit den experimentell beobachteten Mustern übereinstimmen", sagt die Leiterin des Forscherteams Bianca Hermann. "Diesen Ansatz wollen wir nun auf andere Oberflächensymmetrien ausdehnen. Schon jetzt könnten viele Anwendungen in der molekularen Elektronik, Sensorik, Katalyse und Photovoltaik von unserem Modell profitieren. Denn wenn Oberflächenstrukturen besser vorhergesagt werden, können die molekularen Komponenten schon in der Synthese gezielter optimiert werden."
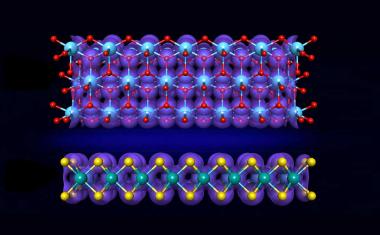
Abb.: Eine Kombination aus statistischer Physik und Simulationen der molekularen Mechanik zusammen mit rastertunnelmikroskopischen Aufnahmen soll die molekulare Selbstorganisation optimieren. (Bild: B.A. Herrmann, WMI, Garching)
Wenn die Natur ihr eigener Ingenieur ist, ordnen sich Moleküle selbst zu komplexen Strukturen an - als Voraussetzung für die Bildung von Membranen, Zellen und anderen molekularen Systemen. Dieses ressourcenschonende Konzept liefert aber auch in der molekularen Elektronik, Sensorik, Katalyse und Photovoltaik funktionale Oberflächenstrukturen. Bei deren Herstellung werden die molekularen Komponenten auf ein Trägermaterial, das Substrat, aufgebracht und finden dann selbst ihren Platz im angestrebten molekularen Netzwerk. Je nach Anwendung zeigen die Bausteine spezifische elektronische, katalytische, sensorische oder photovoltaische Eigenschaften. Die Optimierung dieser hochfunktionellen Molekülschichten hängt aber noch von "trial and error" ab und ist deshalb anspruchsvoll und zeitaufwendig.
In der Zusammenarbeit der Arbeitsgruppen von Bianca Hermann, Thomas Franosch und Erwin Frey im Exzellenzcluster "Nanosystems Initiative Munich" (NIM) gelang nun die Entwicklung eines vereinfachten Modells der molekularen Wechselwirkungen. Dieser Ansatz beruht auf statistischer Physik in einer sogenannten Monte-Carlo-Methodik und detaillierten Simulationen der molekularen Mechanik zusammen mit hochaufgelösten rastertunnelmikroskopischen Aufnahmen. Die Forscherin Marta Balbás Gambra hat in diesem theoretischen Modell Hunderte von Molekülen am Computer zufällig ausgerichtet, um einen Anfangszustand zu simulieren. Diese schematischen Molekülkörper werden dann - in der Berechnung - energetisch angeregt, um Muster zu bilden.
So kann eine - im Vergleich zur Natur - außergewöhnlich große Vielzahl an Mustern erzeugt werden, die in hoher Übereinstimmung auch bei den Experimenten mit dem Rastertunnelmikroskop gefunden werden. "Wir haben sogar ein Muster zunächst theoretisch vorhergesagt und später mit dem Rastertunnelmikroskop nachgewiesen", berichtet der Forscher Carsten Rohr. Nach der Thermodynamik der Physik streben alle Systeme danach, den energetisch günstigsten Zustand einzunehmen. Experimentell wurde gezeigt, dass sich die molekularen Muster ineinander umwandeln können - bis überwiegend nur eine Art "Reifenspurmuster" vorliegt. Dessen günstige energetische Bilanz wurde ebenfalls richtig mit dem "Monte-Carlo-Ansatz" vorhergesagt.
"Letztlich konnten wir zeigen, dass sehr elementare geometrische Eigenschaften der Moleküle diese Vielzahl an Mustern codieren", erklärt der Theoretiker Franosch. "Wir wollen unseren Ansatz nun auch auf andere Substrate, also Oberflächen anderer Symmetrie, ausdehnen. Bereits jetzt aber ist unser Modell ein wichtiges theoretisches Werkzeug, weil es funktionale Oberflächenstrukturen vorhersagen hilft. Damit können die Moleküle schon während der Synthese optimiert werden, um die gewünschten Eigenschaften zu zeigen," erläutert Hermann. Die Physikerinnen und Physiker des Teams, die in unterschiedlichen Fachgebieten arbeiten und ihr Fachwissen hier zusammenführen konnten, sehen Anwendungen im Bereich der molekularen Elektronik, Sensorik, Katalyse und Photovoltaik, auch bei anderen molekularen Wechselwirkungen und bei nur teilweise bedeckten Oberflächen.
Ludwig-Maximilians-Universität München/KP